 First-principles calculation of intrinsic defect chemistry and self-doping in PbTe(第一性原理计算PbTe的内在缺陷化学和自掺杂)
First-principles calculation of intrinsic defect chemistry and self-doping in PbTe(第一性原理计算PbTe的内在缺陷化学和自掺杂)
Anuj Goyal, Prashun Gorai, Eric S. Toberer & Vladan Stevanović
npj Computational Materials 3:42 (2017)
doi:10.1038/s41524-017-0047-6
Published online:10 October 2017
Abstract| Full Text | PDF OPEN
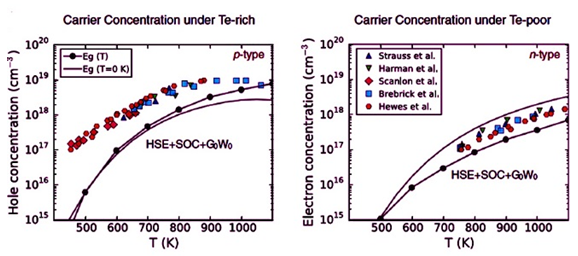
摘要:半导体掺杂本质上受限于其内在缺陷化学。在许多热电材料中,由于强烈的自旋-轨道相互作用导致的窄带隙使固有缺陷化学和自掺杂的原子层次的精确预测变得充满挑战。本研究采用不同层级的理论来模拟PbTe中的点缺陷,将计算结果进行相互类比和对比,并与大量实验数据比对。结果发现,要精确地再现PbTe的固有缺陷化学和已知的自掺杂行为,就必须1)超越密度泛函理论的半局部GGA近似,2)包含自旋-轨道耦合,和3)利用多体GW理论来描述单个带边位置。包含自旋-轨道耦合的杂化HSE函数与G0W0带边偏移的组合,是唯一无需实验数据输入,既可以精确捕获PbTe中随着合成条件的变化导致的本征导电类型,又可以测定载流子浓度的方法。本研究结果再次确认了单个带边位置在缺陷计算中的关键作用,并展示在这些具有挑战的窄带隙材料中可以准确地预测掺杂能力。
Abstract:Semiconductor dopability is inherently limited by intrinsic defect chemistry.In many thermoelectric materials, narrow band gaps due to strong spin–orbit interactions make accurate atomic level predictions of intrinsic defect chemistry and self-doping computationally challenging.Here we use different levels of theory to model point defects in PbTe, and compare and contrast the results against each other and a large body of experimental data. We find that to accurately reproduce the intrinsic defect chemistry and known self-doping behavior of PbTe, it is essential to (a) go beyond the semi-local GGA approximation to density functional theory, (b) include spin–orbit coupling, and (c) utilize many-body GW theory to describe the positions of individual band edges. The hybrid HSE functional with spin–orbit coupling included, in combination with the band edge shifts from G0W0 is the only approach that accurately captures both the intrinsic conductivity type of PbTe as function of synthesis conditions as well as the measured charge carrier concentrations, without the need for experimental inputs. Our results reaffirm the critical role of the position of individual band edges in defect calculations, and demonstrate that dopability can be accurately predicted in such challenging narrow band gap materials.
Editorial Summary
Thermoelectrics: Atomic-level modeling(热电:原子层次上的建模)
PbTe中缺陷的精确建模,需要结合自旋-轨道耦合杂化函数和Green函数理论。热电材料(可将热转换为电能,反之亦然)的掺杂对此类材料的改进来说至关重要;然而,大多数建模方法在原子级别都以失败告终,特别当存在自旋-轨道耦合效应能改变能带位置时。在本研究中,Stevanovic等采用基于第一性原理的密度泛函理论,准确地再现了PbTe的电子结构。他们证实,能与实验结果一致、可为内在缺陷化学建模的唯一方法,是将自旋-轨道耦合的杂化函数(基于屏蔽的库仑势)与通过G0W0近似计算的带边偏移(计算描述扩展系统激发态属性的Green函数理论)相结合。这种层次的理解,对PbTe中掺杂的预测是非常必要的。
Accurate modeling of defects in PbTe lies on the combination of hybrid functionals with spin-orbit coupling and Green function theory. Dopability of thermoelectric materials (that can convert thermal into electrical energy and vice versa) is crucial for their improvement; however most modeling approaches fail on an atomic level, especially if spin-orbit coupling effects (that modify the band position) are present. In this work, Vladan Stevanovic and coauthors manage to accurately reproduce the electronic structure of PbTe with first-principles based density functional theory. They prove that the only approach that can model the intrinsic defect chemistry, in agreement with experimental results, is the combination of hybrid functionals (based on a screened Coulomb potential) with spin-orbit coupling and the band edge shifts calculated through the G0W0 approximation (which calculates the Green function theory that models excited-state properties of extended systems). Such level of understanding is necessary to predict the dopability of PbTe.

 沪公网安备 31010502006565号
沪公网安备 31010502006565号