
氧空位介导的串联催化实现草酸向乙醇酸的电化学转化

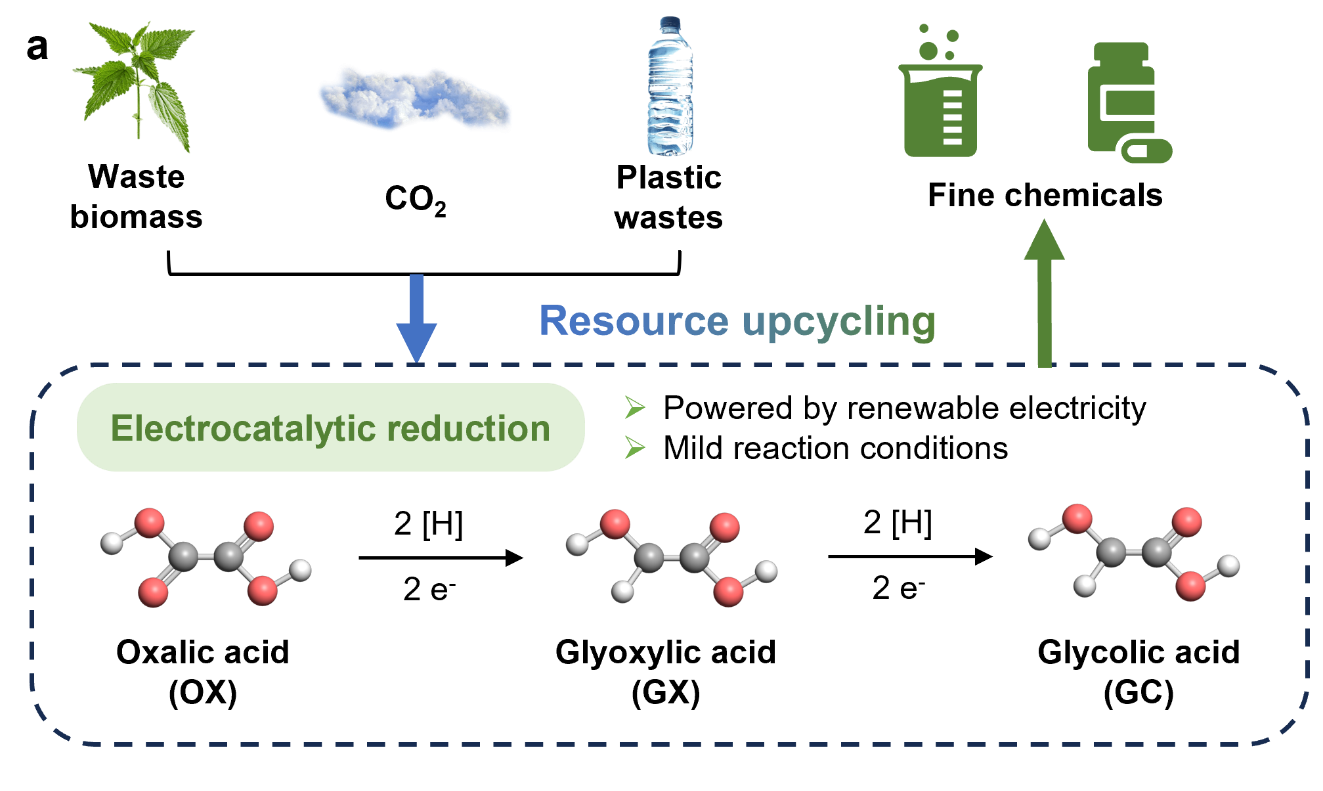
草酸(OX)到乙醇酸(GC)的电化学转化为生物质增值提供了一条可持续的途径,但面临低效的质子耦合电子转移和竞争性析氢的问题。近年来,TiO2由于其优异的稳定性和可调节的电子结构,也被用作草酸加氢反应中的催化剂载体或直接催化剂。特别是,锐钛矿型TiO2在OX氢化方面表现出出色的性能:其3d轨道与OX中C=O基团的孤对电子强烈杂化,促进电子转移;同时,其耐酸特性确保了酸性介质中的结构稳定性。尽管如此,由于反应物和中间体在单个活性位点的竞争吸附,在深度氢化中实现高选择性仍然具有挑战性。
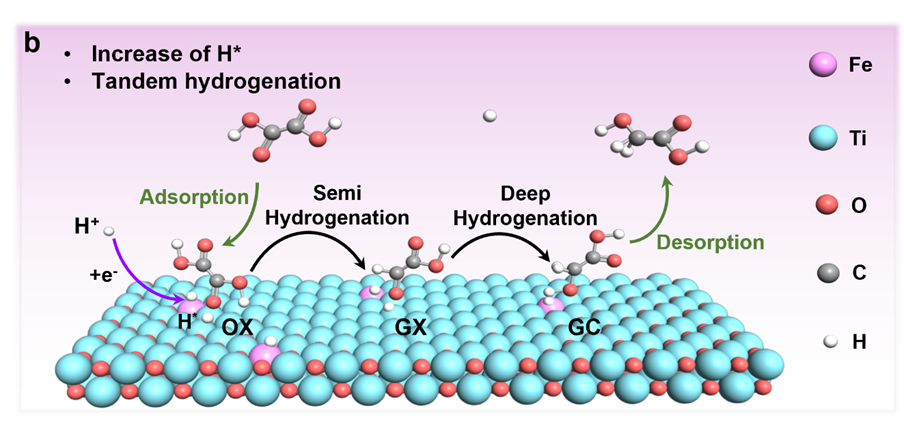
图1:催化机理示意图
过渡金属铁(Fe)具有良好的负吉布斯自由能ΔGH*,可优化质子吸附,而 Fe 掺杂可诱导底物活性位点的重建,调节d带位置和电荷分布以建立协同催化机制。我们假设Fe和Ti的原子级整合可以创建一个双位点串联系统,其中Feδ-位点动态稳定H*中间体(抑制HER),而相邻的Ti3+位点通过电子注入激活C=O键。这种协同作用有效地消除了单中心催化剂固有的竞争吸附。我们通过氧空位(OV)介导的界面工程实现了这种设计,制造了具有Feδ-−OV-Ti3+基序的Fe-TiOX/TP催化剂。原位傅里叶变换红外(FTIR)和密度泛函理论(DFT)计算揭示了串联机制:(i) Feδ-位点加速H*的生成,(ii)Ti3+位点将速率决定步骤(RDS)(HOOCCOOH* → HOOCCO*)的能垒从2.26 eV降低到2.01 eV。
这种协同作用使得GC在工业相关电流密度(50至150 mA cm−2)下的FE达到74.3%,在宽电位窗口[−0.8至−1.2 V vs. RHE]上具有>60%的选择性。该催化剂的60小时稳定性源于电荷补偿的Feδ--OV-Ti3+对,可防止过度还原。
我们的工作建立了设计电化学有机转化中合作活性中心的范例,将原子尺度的洞察力与实际性能相结合。
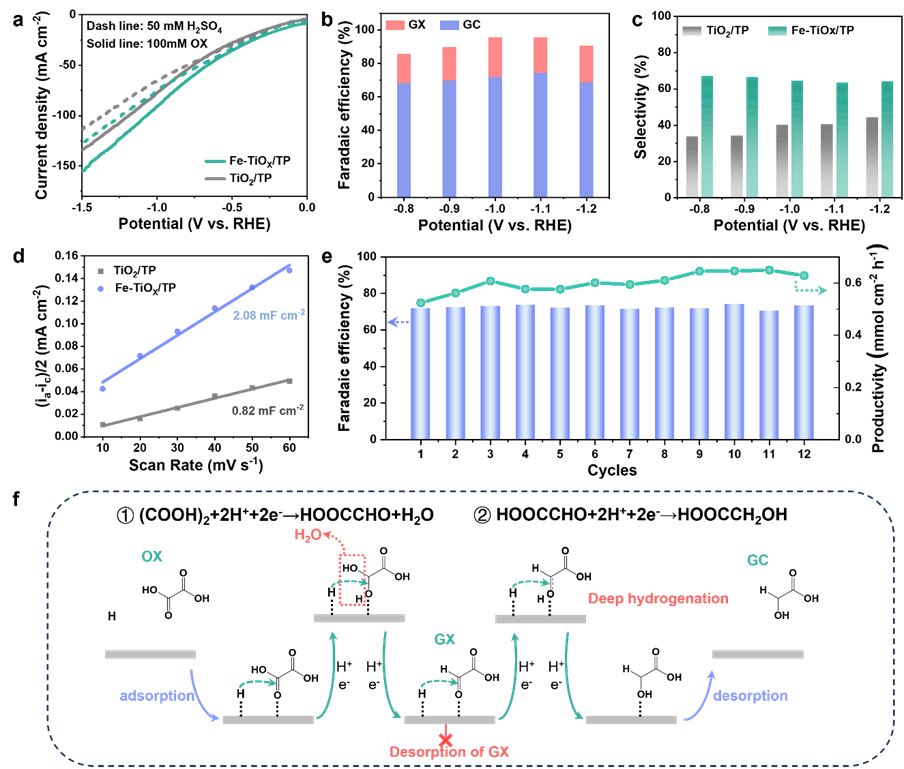
图2:催化性能及反应机制
该研究工作以“Electrochemical conversion of oxalic acid to glycolic acid via oxygen vacancy–mediated tandem catalysis”为题发表在”Science Advances”上(Sci. Adv. 12, eaeb1911 (2026),doi/10.1126/sciadv.aeb1911)。第一作者为2023级硕士生李敏,通讯作者为崔香枝研究员。研究工作得到了国家自然科学基金的资助和支持。
论文链接:https://www.science.org/doi/10.1126/sciadv.aeb1911

