
中国科学院上海硅酸盐研究所刘建军研究员带领的科研团队多年来聚焦“计算电化学”设计电化学储能材料研究领域,形成计算局域结构(表/界面结构、配位结构)电荷转移能力表征电化学活性的特色方向,结合“材料基因”的理念设计高性能电化学储能材料。近日,该科研团队在“计算电化学”设计锂氧气电池正极材料和金属有机电极材料领域取得系列进展。
一、能带排列(Band-alignment)策略降低锂氧气电池过电位
尽管经过多年研究取得了一定进展,然而高的过电位问题仍然是限制锂氧气电池实际应用的重要瓶颈,不同研究工作从提高反应动力学速率与电荷转移、抑制副反应等多角度探索,但效果欠佳,归根结底是过电位本征机制还不明确。刘建军研究员带领的科研团队与中国科学院大学刘向峰教授、南方科技大学张文清教授团队合作,全面研究锂氧气电池中Li+/O2的化学脱附和电荷转移动力学对充电电位的关系规律,发现电极材料与Li2O2界面电荷转移决定性地造成高过电位,提出能带排列方法设计电极材料的新策略,计算预测Li2O2在6种正极材料上的过电位与实验测量值取得较好一致性,证明界面电荷转移的重要作用。进一步基于能带排列方法开展高通量计算,筛选出17种充电电压<3.7 V的高活性正极材料,针对3种实验证实的高活性正极材料(IrO2, RuO2, TiC),理论预测的充电电位与实验观测值误差在0.25 V以下,并预测了潜在的具有低充电过电位(<0.5 V)的正极材料MnN和Cr2O3。
该工作指出固-固相界面电化学反应中,界面电荷转移过程是影响能量转化效率的关键因素,提出的能带排列策略可以进一步推广到其他固-固相界面电化学电池,如Li-CO2电池、Li-S电池等。研究成果发表在Energy Environ Sci杂志上,第一作者是上海硅酸盐所王有伟助理研究员。
文章信息:Energy Environ. Sci. 2020, DOI: 10.1039/D0EE01551B
二、高能量密度的金属有机电极材料
金属有机电极材料具有原料丰富、结构柔性、易于降解、环境友好等优势,然而电化学活性仅仅局限在金属阳离子位点造成能量密度低的问题,实现金属阳离子与有机结构协同电化学至关重要。刘建军研究员带领科研团队通过利用金属有机材料中TM-X(X=O,F,S…)的强共价杂化作用,以及有机官能团-NO2,-N=N-,-C=O的高还原势,设计了一类具有“阴-阳离子协同电化学”特征的“多级化学键和多活性位点”的高能量密度金属有机电极材料[Cu2(p-O2NC6H4CO2)4(EtO)2],8电子转移过程取得了697 Wh/kg的能量密度,高于当前已报道的具有最高能量密度680 Wh/kg的CuTCDQ。[Cu2(p-O2NC6H4CO2)4(EtO)2]中Cu-O的强共价杂化造成Cu-O阴阳离子协同电化学,提高了材料的放电电压(3.66~2.08 V),同时,官能团-NO2与Li+的嵌入反应电压高于析出反应Cux+(0<x<1)→Cu0+而优先提供活性位点,因此具有高比容量(243 mA h g-1)优势。研究成果发表在ACS Energy Lett上,第一作者是2019级博士研究生赵晓琳。
文章信息:ACS Energy Lett. 2020, 5, 477-485, DOI: 10.1021/acsenergylett.9b02630
以上研究成果得到了国家自然科学基金面上项目、上海市科委国际合作、上海市“扬帆计划”等项目资助。
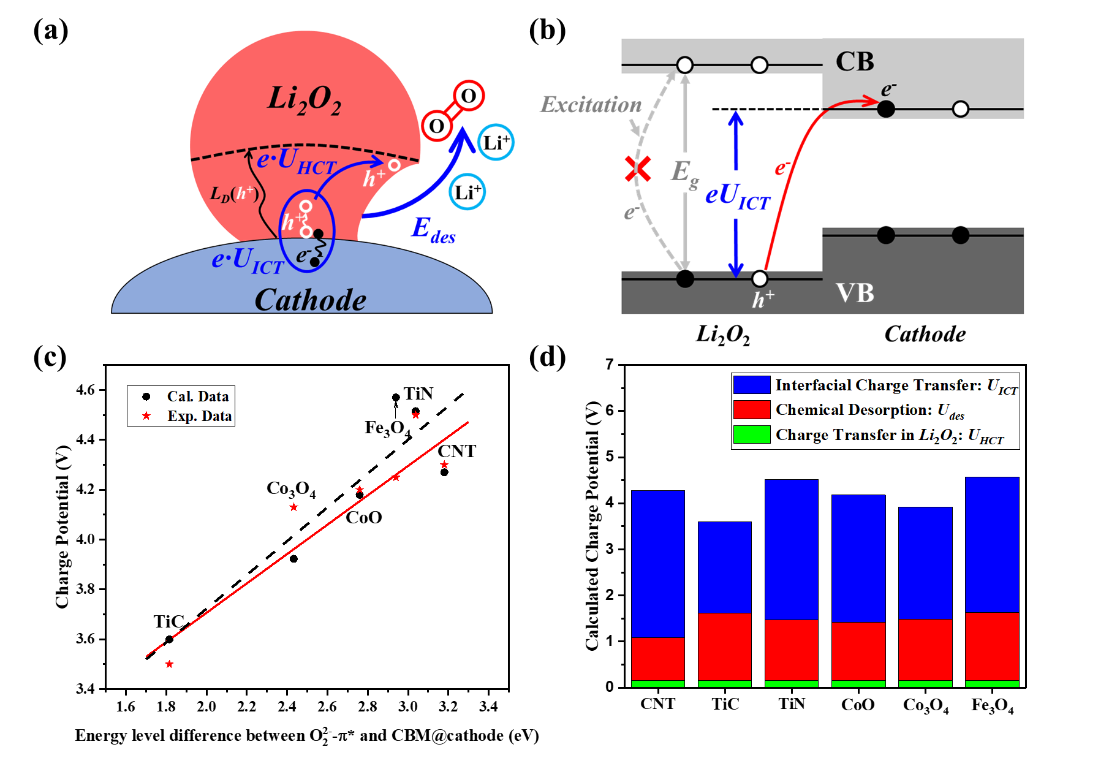
(a) Li2O2分解过程,包括化学脱附(e·Udes)和在Li2O2内部的电荷传输(e·UHCT)以及在Li2O2@正极界面的电荷转移(e·UICT);(b)能带排列(Li2O2中O22--π*能级与正极导带底能级的匹配关系)策略示意图;(c)充电电位计算值和实验值对比图;(d)化学脱附过程、电荷转移过程对充电电位的贡献占比。
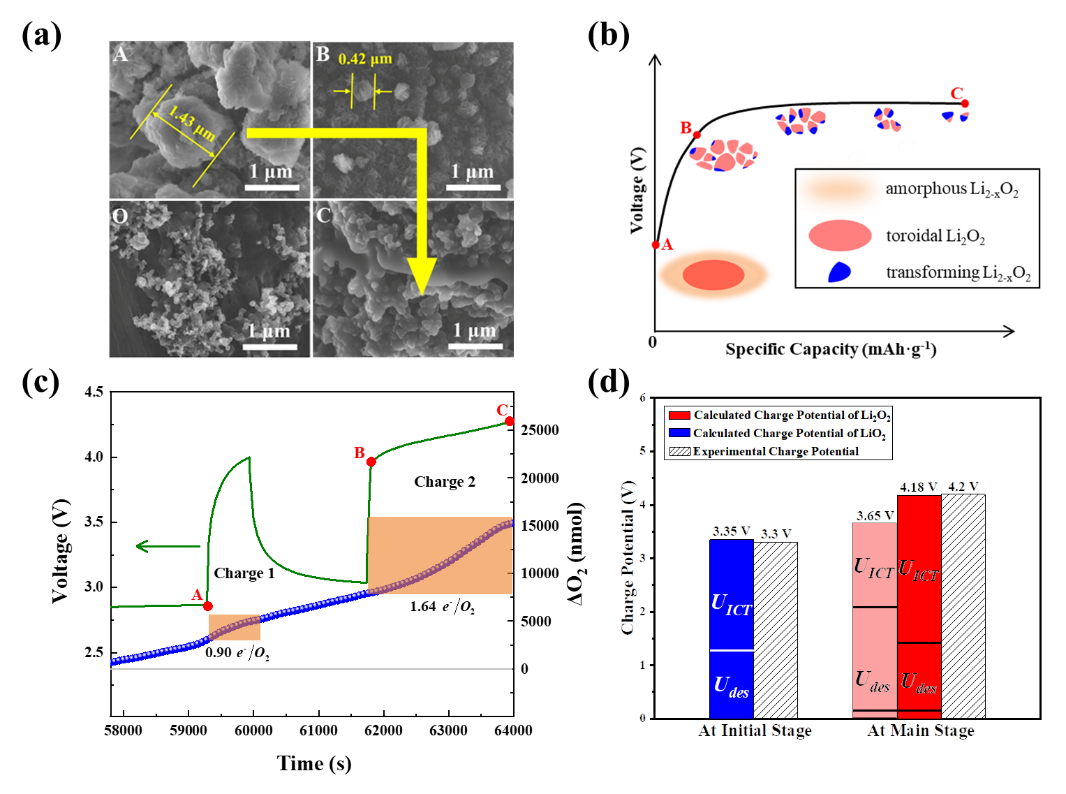
(a)完全放电(A)、充电至4 V时(B)、完全充电(C)的Li2O2@CoO和纯CoO正极(O)的SEM图像;(b) Li2O2@CoO的形貌变化示意图;(c)在充电电压低于4 V和高于4 V且中止0.5 h的情况下,Li2O2@CoO的DEMS、电压曲线;(d) LiO2和Li2O2分解的充电电位计算值与实验值的对比图。

高通量计算筛选高活性正极材料。(a)化学脱附过程对充电电位贡献关系;(b)界面电荷转移势垒对充电电位贡献关系;(c)能带排列粗略对充电电位的关系规律;(d)筛选的充电电位<3.7 V的高活性正极材料,并将其与TiC、MnO2、RuO2和IrO2实验获得数据对比。
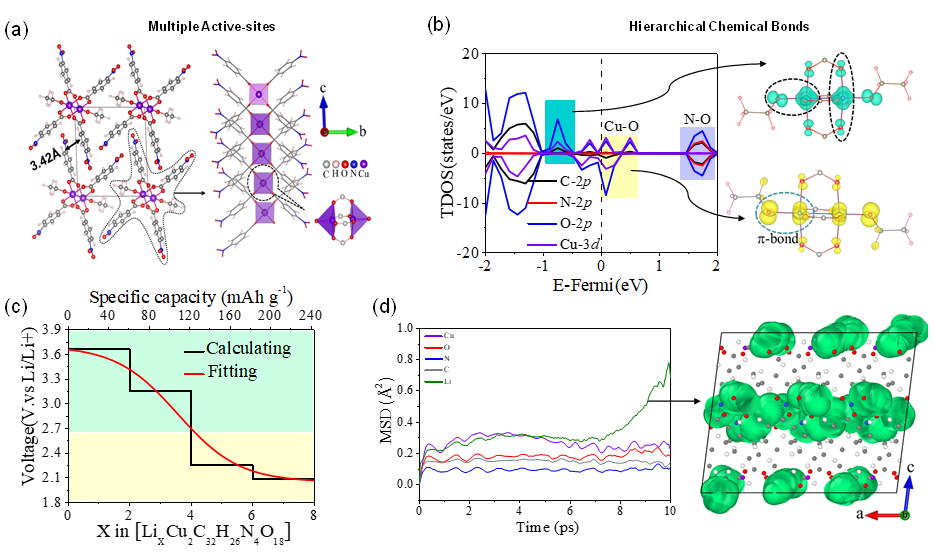
[Cu2(p-O2NC6H4CO2)4(EtO)2]的(a)晶体结构和(b)多级化学键的电子结构 (c)电压曲线和理论比容量 和 (d)锂离子迁移路径以及MSD的动力学计算。

